Noen ganger er det ikke opplagt om barnet ble en jente eller en gutt. Ivaretakelse av pasienter med usikkert kjønn i Norge, er organisert som en flerregional behandlingstjeneste lokalisert til Haukeland Universitetssjukehus og Oslo Universitetssykehus. I denne artikkelen vil vi å gi en kort beskrivelse av tilstander, utredning og behandling – eksemplifisert ved noen kasuistikker.
Tekst: Hilde Bjørndalen, Oslo Universitetssykehus, Anne Wæhre, Oslo Universitetssykehus, Birgitta Ivarson, Haukeland Universitetssjukehus og Helge Ræder, Haukeland Universitetssjukehus
Rundt 1 av 1000-4500 barn fødes med genitale variasjoner som innebærer en somatisk forstyrrelse eller variasjon i kjønnsutviklingen (1). Usikker somatisk kjønnsutvikling forkortes gjerne DSD (disorders of sexual development). Denne artikkelen belyser ikke diagnosen transseksualisme, der en person oppfatter å ha en kjønnsidentitet som ikke samsvarer med ytre genitalier. DSD er alltid medfødt og skyldes genetiske og intrauterine forhold som påvirker gonader (=anlegg for enten eggstokk eller testikler) og/eller genitalia. Resultatet er at kjønnet ved fødsel ikke kan fastsettes umiddelbart, eller at det senere i livet fremkommer tegn på variasjoner i kjønnsdifferensieringen, f. eks ved pubertet eller ved forventet pubertet.
Når et barn blir født med uklare kjønnskarakteristika er det en situasjon som krever rask og grundig informasjon av foreldrene, hurtig målrettet utredning og fastslåelse av oppdragerkjønn, samt godt forberedt behandling og langtidsoppfølging. Det er nødvendig med tverrfaglig kunnskap i team med bl.a. barneendokrinolog, barnekirurg, plastikkirurg, gynekolog, barnepsykiater eller -psykolog og sykepleier.
Viriliseringsgrad og Müllerstrukturer
I nyfødtperioden gir DSD seg typisk til kjenne som virilisert klitoris eller proximal hypospadi avhengig av viriliseringsgrad og karyotype. Mekanismene er knyttet til undervirilisering med for lite testosteron fra testiklenes Leydigceller eller androgenoverskudd med androgener fra andre kilder (feks binyrene ved medfødt binyrebarksvikt eller fra placenta ved aromatasemangel). Viriliseringsgraden gir et spekter fra sammenvoksning av de store kjønnsleppene baktil via bifid scrotum til normal scrotum. Noen tilstander opptrer asymmetrisk med store kjønnslepper på den ene siden og hemiscrotum på den andre siden.
Müllerstrukturene gir opphav til livmor og tuber, og tilbakedannes normalt ved 46,XY-karyotype under AMH (anti Müllerisk hormon)-påvirkning fra testiklenes Sertoli-celler. Ved enkelte 46,XY-DSD-tilstander (gonadedysgenesier), ses gjenværende rester av Müllerstrukturer, f.eks i form av liten uterus, hemiuterus eller prostatisk utriculus.
Klassifisering
DSD klassifiseres i tre undergrupper: 46,XX-DSD, kjønnskromosom DSD og 46,XY-DSD. Den vanligste tilstanden finner vi i gruppen 46,XX-DSD med androgenoverskudd, nemlig 21-hydroksylase-svikt. Den vanligste kjønnskromosom DSD-tilstanden vi ser er mixed gonadal dysgenesi med karyotypen 45,X0/46,XY eller varianter av denne. 2/3 av disse pasientene vokser opp som gutter, resten jenter. Hos 46,XY-DSD-tilstandene vil noen vokse opp som jenter, de fleste som gutter. Hvis vi finner Müllerstrukturer og reduserte AMH-verdier, foreligger som regel en gonadedysgenesi (som rammer både Leydigcellene og Sertolicellene). Det finnes også 46,XX-gonadedysgenesier. Hvis vi ikke finner Müllerstrukturer, foreligger sannsynligvis androgensyntese-defekt eller androgen-reseptor-resistens (46,XY-DSD med undervirilisering som kun rammer Leydigcellene).
Vi presenterer i det følgende fire kasuistikker som belyser håndteringen av disse pasientene.
Congenitt adrenal hyperplasi
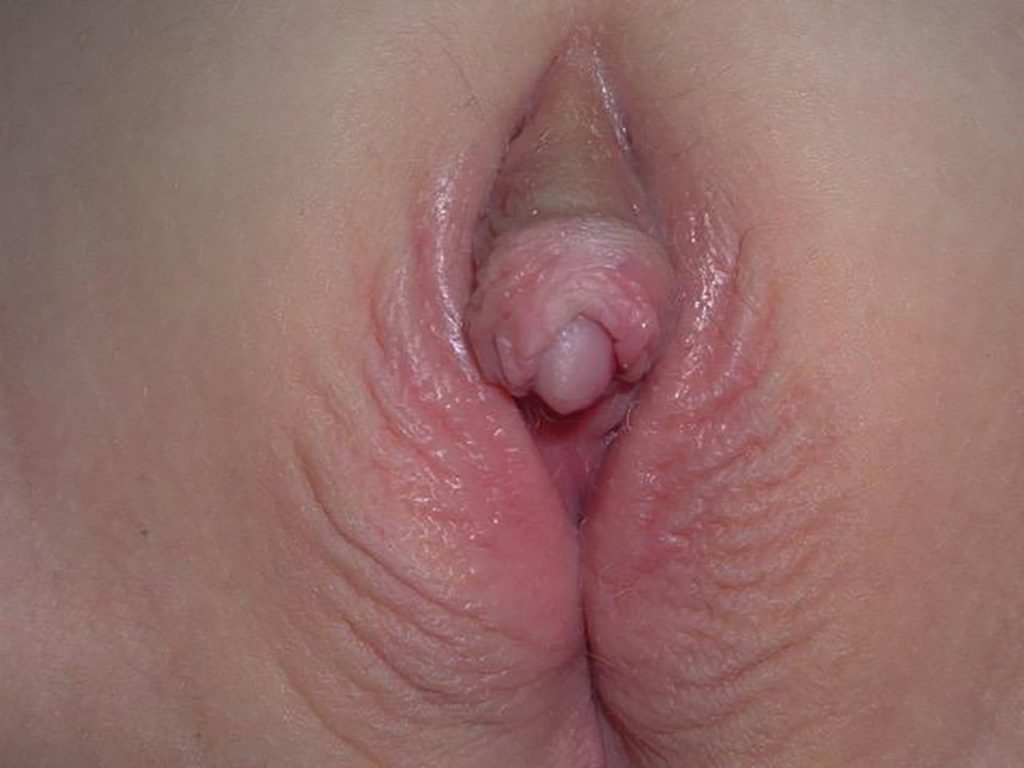
Bildet viser scrotalisert hud, utrethra munner på tuppen av genitaltubercelen. Bildet er ikke fra pasienten i kasuistikken.
Terminfødt baby innlagt ved 7 dagers alder pga slapphet og gulpetendens. Blodprøvene viste Na 122 nmol/l, K 9,4 mmol/l, Blodsukker 1,9 mmol/l, TSH 6,2, FT4 23,8, Testosteron 37,1, cortisol 655, aldosteron > 4162, 17- OH-progesteron 3000, ACTH 135. Kromosomundersøkelse 46,XX.
Diagnose: Virilisert jente med 21-hydroxylasesvikt – congenitt adrenal hyperplasi.
Behandling: Natrium-tilskudd, Cortisonacetat og Florinef.
Operert med feminiserende genitalplastikk ved 6 måneders alder, reoperert 7 år senere pga fistel mellom urethra og vagina.
Etter at Nyfødtscreeningen ble utvidet i 2012 blir pasienter med tilstanden oftest diagnostisert før de utvikler elektrolyttforstyrrelser. Den medikamentelle behandlingen kommer derfor raskt i gang. Når det gjelder den feminiserende kirurgiske behandlingen har trenden vært at dette utsettes sammenlignet med tidligere, basert både på økende kunnskap om kompleksiteten bak psykoseksuell utvikling, som formes etter en lang prosess påvirket av gener, kjønnsforskjeller i hjernestruktur, prenatal androgen eksponering i tillegg til sosiale og kulturelle faktorer. Et argument for å utsette operasjon er pasientens mulighet til å være med å bestemme dette selv. Kirurgi for å sikre så normal blærefunksjon som mulig må ofte gjøres tidligere. Foreldrene blir grundig informert om hva som må gjøres umiddelbart og hva som kan/bør utsettes. Forskning viser at pasienter med diagnosen CAH som da oppdras som jenter kan ha en tradisjonelt mer typisk mannlig oppførsel, men majoriteten (95 %) utvikler en kvinnelig kjønnsidentitet (3). Det lavere antallet av kvinner som har en mannlig identitet er ikke nødvendigvis de som har mest maskuliniserte genitalier ved fødsel, men der man har en forsinket diagnostisering og kraftig virilisering kan man ha behov for en mer forsiktig tilnærming i det tverrfaglige teamet med psykologisk støtte til foreldre og diskusjon rundt fremtidig kjønnsidentitet. (4)
Mixed gonadal dysgenesi
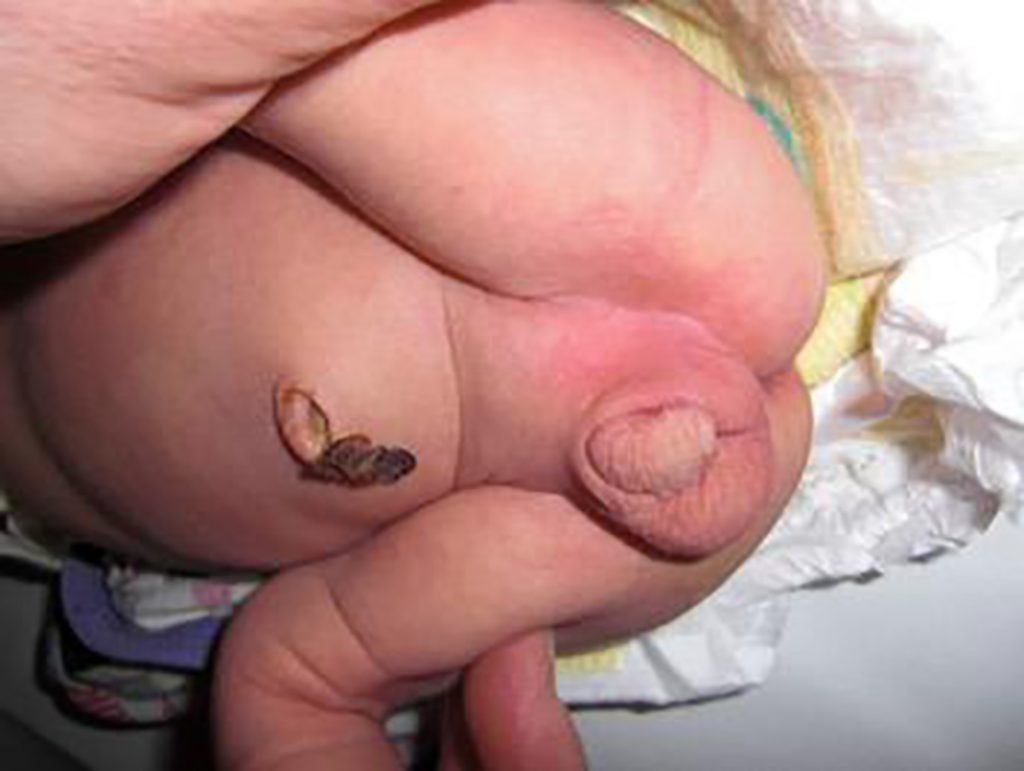
Bildet viser blant annet asymmetriske labioscrotalfolder med mindre grad av scrotalisering på venstre side hos pasient med miksed gonadal dysgenesi.
Pasienten var mors andre barn. I svangerskapet påvist nakkefold. Barnet ble født i uke 38, indusert pga vekstretardasjon, så keisersnitt pga truende asfyksi. Det var Oligohydramnion, Apgar 9/10, FV 2280, FL 44 cm, bortsett fra genitalia normal organstatus og nevrologisk status. Ved undersøkelse av genitalia externa fant man på høyre side scrotalisert labioscrotalfold med gonade som på ultralyd så testikulær ut. På venstre side så man store kjønnsleppe-aktig labioscrotalfold uten gonade, og man så heller ikke ved ultralyd noen gonade i abdomen. Videre så man penoscrotal hypospadi og fallos med strukket lengde 2.0 cm. Billeddiagnostikk av abdomen (ultralyd, kontrastundersøkelser) påviste ingen tegn til Müllerstrukturer, videre så man en urogenital sinus ca 2.5 cm lang og ingen tegn til forstørrede binyrer.
Blodprøver viste testosteron 1.8 nM, Androstenedion 0.8 nM, AMH 275 pM. Det var normal blod-glucose og normale repeterte urin-elektrolytter, lavnormale 21OH og 11DOC. Hurtigtest viste Y-kromosomale gener (SRY og amylogenin).
Vi konkluderte med at barnet burde vokse opp som gutt basert på tilstedeværelse av Y-kromosomale gener, god Sertolifunksjon (dvs. ikke Müllerstrukturer og AMH 275 pM) og tilstrekkelig god Leydigfunksjon (penoscrotal hypospadi, fallos ca 2 cm, en testikkel i scrotalisert labioscrotalfold, basal testosteron 1.8 nM dag 3 og Androstenedion 0.8 nM dag 3). Såpass grad av virilisering predikerer en godt virilisert hjerne og således lav sannsynlighet for senere kjønnsinkongurens (opplevelse av å ha en identitet som ikke samsvarer med den identiteten man vokser opp som). Asymmetrien av labioscrotalfoldene taler for enten mixed gonadal dysgenesi eller ekte hermafroditisme.
Kromosomsvaret 45,X0/46,XY-mosaikk bekreftet mixed gonadal gonadedysgenesi med 9/30 45,X0-cellelinjer. Barnet fikk korrigert sin hypospadi i to seanser ved henholdsvis 1 års alder og 1.5 års alder. Siden tilstanden er genetisk beslektet med Turners syndrom, la vi opp til en utredning også med tanke på venstresidige hjerteanomalier, nyreanomalier, mediaotitter, thyroiditt, hypertensjon, ortopediske problemer, lengdevekst (veksthormonbehov), pubertet (testosteron-substitusjon). Det ble også gitt veiledning ved avdeling for medisinsk genetikk. Y-komponent i gonadene kan disponere for dysgerminomer, og vi fulgte derfor gonadene med klinisk undersøkelse av testikkelen som lå i pungen, årlig ultralyd og tumormarkører (total beta hCG, AFP og LDH). Den abdominelle gonaden ble påvist med laparoskopi og senere fulgt billeddiagnostisk. Vi la opp til elektiv orkidektomi når gutten var utvokst.
Complete Androgen Insensitivity syndrome
Pasienten ble henvist ved 16 års alder på grunn av primær amenoré. Operert for lyskeebrokk ved 1 års alder, ellers tidligere frisk. Brystutvikling fra 12 års alder.
Blodprøver: FSH 4,9 IU/l, LH 22,6 IU/l, østradiol 0,1 nmol/l, testosteron 18,2 nmol/l, kromosomundersøkelse viste 46,XY. Videre genetisk utredning viste sykdomsgivende mutasjon i genet for androgenreseptor.
MR lille bekken: To rudimentære uterinanlegg, gonader beliggende på ovarienes plass.
Diagnose: CAIS – complete androgen insensitivity syndrome.
Androgen insensitivitetssyndrom kan være enten komplett eller partielt, avhengig av mutasjonen i androgenreseptor-genet. Ved komplett androgen insensitivitet er pasienten fenotypisk jente/kvinne, og diagnosen stilles derfor ofte som i dette tilfelle i forbindelse med utredning av primær amenoré. Det er en sjelden tilstand med forekomst på 1: 10 000 – 20 000.
Gonadene som kan ligge intra-abdominalt eller i lyskene produserer testosteron under påvirkning av LH, men testosteronet virker ikke – verken i CNS eller den somatiske kjønnsutviklingen. Testosteron omdannes til østrogen ved hjelp av enzymet aromatase og pasientene har derfor en kvinnelig kjønnsidentitet, og kvinnelige kroppsproposjoner med helt normal brystutvikling og feminine ytre genitalia. Uterus kan mangle helt eller være rudimentær, vagina er ofte lite utviklet. Det er vanligvis androgenreseptorer i hud som er ansvarlig for axille- og pubes-behåring, samt akne og fet hud som oppstår i forbindelse med pubertet. Disse pasientene har derfor lite av dette. Det diskuteres i flere sammenhenger om det er fare – og eventuelt hvor stor – for kreftutvikling i intraabdomialt beliggende gonader. Når gonadene er fjernet, trenger pasientene østrogentilskudd.
Mutasjoner i androgen-reseptor-genet som gir partiell androgen insensitivitet er sjeldnere og kan fører til et bredt spekter av tilstander fra en virilisert jente til undervirilisert gutt, eventuelt infertilitet hos en ellers fenotypisk helt normal mann.
Proximal hypospadi
Barn forløst med elektiv sectio (grunnet stort barn og seteleie) uke 39+0, vekt 5440g. Overflyttet Haukeland Universitetssykehus grunnet uklare ytre kjønnskarakteristika.
Friske foreldre, ukomplisert svangerskap. Mors søster var født med en problemstilling med uklare kjønnskarakteristika og er blitt kirurgisk og hormonelt behandlet som jente.
Funn ved innkomst: Relativt mangelfullt utviklet fallos, palpable gonader i ”labioscotalfold” . Meatus beliggende perinealt. Ved ultralyd påvises ikke Müllerstrukturer og karyotypen er 46, XY. Utredning senere påviser partiell androgen insensitivitet. Hans bilaterale kryptorkisme blir behandlet med orkidopeksi. Pasienten vurderes av plastikkirurger og man finner indikasjon for operativt inngrep i to seanser (Ved OUS er en seanse vanlig), først ved 1 års alder og neste inngrep ved 1,5 års alder. Det blir gitt testosteronbehandling intramuskulært (25 mg månedlig i 3 måneder) mellom inngrepene da penis er liten og der er lite vev for å gjøre kirurgien teknisk lettere.
Ved seanse 1 ble vev flyttet fra forhuden over på undersiden av penis. Det forelå svært korte ventrale strukturer og en penis som nærmest var loddet ned mot perineum, mens scrotum var ufusjonert. Under operasjonene deles urethralplaten og stramme drag ventralt eksideres for å få rettet ut penis. Pga restkrumming på ca 45 grader satte man en oppstrammende sutur dorsalt.
Ved seanse 2 ble det laget en ny uretra ved å ”tubulere” forhudsvevet som ble flyttet i seanse 1. Man skar ut en hudbro fra perinealt beliggende meatus frem til tuppen av glans. Hudbroen ble tubulert rundt et 8.0 babyfeedkateter. Scrotum ble løst ut på begge sider av penis og transponert til undersiden av penis.
Ved 4 års-kontrollen forelå restkrumming på 45 grader som man fulgte konservativt til 11 års alder. Han hadde ingen UVI-episoder, men måtte sitte og tisse, forøvrig uproblematisk vannlating. Det blir så gjort en utrettende prosedyre på penis som inkluderte bruk av munnslimhinne for å reparere en uretradefekt. Seks måneder senere hadde slimhinnegraftet på penis blitt mykt og ”modent” og det ble gjort tubulering rundt et 12 kateter. Etter dette var penis rett, han kunne stå å tisse, og videre forløp var ukomplisert.
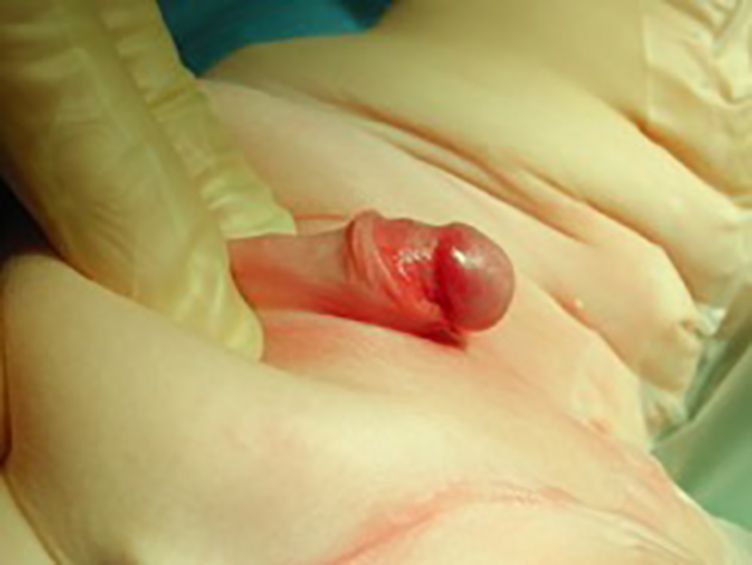
Hypospadi med relativt mangefult utviklet fallos.
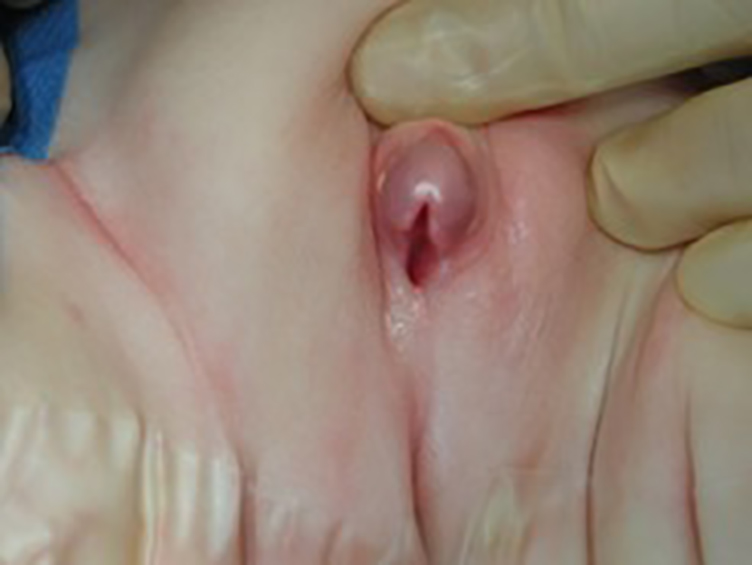
Bildet viser hypospadi der meatus ligger perinealt.
Hva skal du gjøre når kjønnet er uklart?
Kontakt behandlingstjenesten i Bergen eller Oslo så tidlig som mulig. Barnet overflyttes til Nyfødtavdeling og den videre utredningen gjøres der. Kontaktinformasjon og mer detaljer finnes i kapitlet om feil i somatisk kjønnsutvikling i generell veileder i pediatri (kapittel 2.14) (2).
- Grundig familie- og svangerskapsanamnese
- En nøye klinisk undersøkelse som beskriver genitaltuberkel, labioscrotalstruktur, gonadeplasseringen og eventuelle syndromtrekk.
- Bildediagnostikk for å kartlegge de indre genitalia (spør om tilstedeværelse av Müllerstrukturer og lokalisasjon av gonader).
- Blodprøver for karyotype + hurtigtest for Y-kromosomalt materiale, hormonprøvene FSH, LH, testosteron, AMH (anti Müllerisk hormon) og 17-OH-progesteron (som stiger ved 21-hydroksylasesvikt) bestilles som ø.hjelp.
Referanser:
- Hughes, Nihoul-Fekete, Thomas, & Cohen-Kettenis, 2007; Thyen, Lanz, Holterhus, & Hiort, 2006)
- Generell veileder i pediatri (kapittel 2.14)
- Meyer-Bahlburg, 1999; Berenbaum and Beltz, 2011, Hines, 2011a).Horm Behav. 2016 Nov;86:8-20. doi: 10.1016/j.yhbeh.2016.08.008. Epub 2016 Aug 27. Recalled and current gender role behavior, gender identity and sexual orientation in adults with Disorders/Differences of Sex Development. Callens et al) and (Gender identity, gender assignment and reassignment in individuals with disorders of sex development: a major of dilemma. J Endocrinol Invest. 2016 Nov;39(11):1207-1224))
Når et barn blir født med uklare kjønnskarakteristika, er det en situasjon som krever rask og grundig informasjon til foreldrene, hurtig målrettet utredning og fastslåelse av oppdragerkjønn, samt godt forberedt behandling og langtidsoppfølging.